RNASeq workflow for meta analysis with Cvig immunity
Summary
We are analyzing 5 datasets of Eastern Oyster, C. virginica, exposed to Perkinsus marinus infections.
Reference genome
All datasets work with the Eastern Oyster, C. virginica, so I need to download the genome, link2.
cd /mmfs1/gscratch/scrubbed/elstrand/genomes
mkdir C_gigas
wget https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/002/022/765/GCF_002022765.2_C_virginica-3.0/GCF_002022765.2_C_virginica-3.0_genomic.fna.gz
wget https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/002/022/765/GCF_002022765.2_C_virginica-3.0/GCF_002022765.2_C_virginica-3.0_genomic.gff.gz
wget https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/002/022/765/GCF_002022765.2_C_virginica-3.0/GCF_002022765.2_C_virginica-3.0_genomic.gtf.gz
## modifying the empty gene id columns
zcat GCF_002022765.2_C_virginica-3.0_genomic.gtf.gz | awk -F"\t" '{if($9 ~/gene_id ""/ ) gsub(/gene_id ""/,"gene_id \"unknown_transcript_1\"",$9);print $0}' | gzip > mod_GCF_002022765.2_C_virginica-3.0_genomic.gtf.gz
Final script
I was messing with haivng the fastp arguments with $4 but was getting errors with that. So changed the clipping parameters as needed.
rnaseq_Cvir.sh
#!/bin/bash
#SBATCH --account=srlab
#SBATCH --error=/gscratch/scrubbed/elstrand/Cvir_disease_meta/scripts/output/"%x_error.%j" #if your job fails, the error report will be put in this file
#SBATCH --output=/gscratch/scrubbed/elstrand/Cvir_disease_meta/scripts/output/"%x_output.%j" #once your job is completed, any final job report comments will be put in this file
#SBATCH --partition=cpu-g2-mem2x
#SBATCH --nodes=1
#SBATCH --time=1-20:00:00
#SBATCH --mem=200G
#SBATCH --ntasks=7
#SBATCH --cpus-per-task=2
#conda activate nextflow
## paths defined in the sbatch command
samplesheet=$1
output=$2
multiqc_title=$3
# fastp=$4
## paths for all datasets
genome="/gscratch/scrubbed/elstrand/genomes/C_virginica/GCF_002022765.2_C_virginica-3.0_genomic.fna.gz"
gff="/gscratch/scrubbed/elstrand/genomes/C_virginica/GCF_002022765.2_C_virginica-3.0_genomic.gff.gz"
gtf="/gscratch/scrubbed/elstrand/genomes/C_virginica/mod_GCF_002022765.2_C_virginica-3.0_genomic.gtf.gz"
nextflow run nf-core/rnaseq -resume \
-c /gscratch/scrubbed/elstrand/uw_hyak_srlab_estrand.config \
-profile singularity \
--input ${samplesheet} \
--outdir ${output} \
--gtf ${gtf} \
--gff ${gff} \
--fasta ${genome} \
--trimmer fastp \
--extra_fastp_args '--cut_mean_quality 30 --trim_front1 10 --trim_front2 10' \
--aligner star_salmon \
--skip_pseudo_alignment \
--multiqc_title ${multiqc_title} \
--deseq2_vst
How to run (example from dataset #4):
conda activate nextflow
cd /gscratch/scrubbed/elstrand/Cvir_disease_meta/dataset4
sbatch -J Cvir_disease_rnaseq_dataset4 ../scripts/rnaseq_Cvir.sh \
/gscratch/scrubbed/elstrand/Cvir_disease_meta/samplesheets/samplesheet_rnaseq_dataset4.csv \
/gscratch/scrubbed/elstrand/Cvir_disease_meta/dataset4 \
Cvir_disease_rnaseq_dataset4 \
'--cut_mean_quality 30 --trim_front1 0 --trim_front2 0'
How to check output while script is running:
cd /gscratch/scrubbed/elstrand/Cvir_disease_meta/scripts/output
### two log files are generated from each sbatch script
### the titles are based on the -J job name indicated above
Cvir_disease_rnaseq_dataset4_error.22760577
Cvir_disease_rnaseq_dataset4_output.22760577
less <output_file>
## Shift + G to get to the bottom of the file to see something like this:
[ec/008475] NFC<E2><80><A6>ginica-3.0_genomic.fna.gz) | 1 of 1 <E2><9C><94>
[c0/569e4f] NFC<E2><80><A6>ginica-3.0_genomic.gtf.gz) | 1 of 1 <E2><9C><94>
[c0/63882a] NFC<E2><80><A6>virginica-3.0_genomic.fna) | 1 of 1 <E2><9C><94>
[93/224f7b] NFC<E2><80><A6>-3.0_genomic.filtered.gtf) | 1 of 1 <E2><9C><94>
[9a/2ccc66] NFC<E2><80><A6>virginica-3.0_genomic.fna) | 1 of 1 <E2><9C><94>
[d9/a97eb2] NFC<E2><80><A6>virginica-3.0_genomic.fna) | 1 of 1 <E2><9C><94>
[33/76468b] NFC<E2><80><A6>virginica-3.0_genomic.fna) | 1 of 1 <E2><9C><94>
[- ] NFC<E2><80><A6>_SETSTRANDEDNESS:CAT_FASTQ -
[0b/e866d0] NFC<E2><80><A6>P:FASTQC_RAW (SRX18040063) | 61 of 61 <E2><9C><94>
[cc/41ac6d] NFC<E2><80><A6>_FASTP:FASTP (SRX18040033) | 61 of 61 <E2><9C><94>
[26/ac53fa] NFC<E2><80><A6>:FASTQC_TRIM (SRX18040033) | 61 of 61 <E2><9C><94>
[16/899d3d] NFC<E2><80><A6>EX (genome.transcripts.fa) | 1 of 1 <E2><9C><94>
[dc/663c4e] NFC<E2><80><A6>FQ_SUBSAMPLE (SRX18040033) | 61 of 61 <E2><9C><94>
[ce/ab5214] NFC<E2><80><A6>SALMON_QUANT (SRX18040033) | 61 of 61 <E2><9C><94>
[73/e2416c] NFC<E2><80><A6>R:STAR_ALIGN (SRX18040038) | 22 of 62, failed: 1, retries: 1
[43/19aa0b] NFC<E2><80><A6>AMTOOLS_SORT (SRX18040038) | 16 of 21
[f4/a1733e] NFC<E2><80><A6>MTOOLS_INDEX (SRX18040056) | 0 of 16
[- ] NFC<E2><80><A6>TS_SAMTOOLS:SAMTOOLS_STATS -
[- ] NFC<E2><80><A6>SAMTOOLS:SAMTOOLS_FLAGSTAT -
[- ] NFC<E2><80><A6>SAMTOOLS:SAMTOOLS_IDXSTATS -
[a2/5d5395] NFC<E2><80><A6>SALMON_QUANT (SRX18040038) | 16 of 21
[4f/4acd78] NFC<E2><80><A6>RKDUPLICATES (SRX18040056) | 0 of 16
Plus 30 more processes waiting for tasks<E2><80><A6>
How I got there
Notes in chronological order of trial and error.
Datasets
See ABOUT page for table of datasets
| Set # | Species | Data Type | stress class | stressor | Phenotype | Phenotpe Summary | Reference | DOI | SRA IDs | SRA links | meta data file | counts table | DEG data |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | C. virginica | T | disease | Perkinsus | infection tolerance | resilience | Proestou et al. 2023 | https://doi.org/10.3389/fgene.2023.1054558 | SRX180s | https://www.ncbi.nlm.nih.gov/sra?LinkName=bioproject_sra_all&from_uid=894694 | SraRunTable.csv | ||
| 2 | C. virginica | E,T | disease | Perkinsus | infected | sensitivity | Johnson et al. 2020 | https://doi.org/10.3389/fmars.2020.00598 | SRX765s | https://www.ncbi.nlm.nih.gov/Traces/study/?acc=SRP246310&o=acc_s%3Aa | SraRunTable(1).csv | ||
| 3 | C. gigas & virginica | T | disease | Perkinsus | infection tolerance | resilience | Chan et al. 2021 | 10.3389/fgene.2021.795706 | SRX564s | https://www.ncbi.nlm.nih.gov/Traces/study/?acc=SAMN11031730&o=acc_s%3Aa | SraRunTable(2).csv | ||
| 4 | C. virginica | T | disease | Perkinsus | infection | sensitivity | Sullivan and Proestou 2021 | https://doi.org/10.1016/j.aquaculture.2021.736831 | SRX984s | https://www.ncbi.nlm.nih.gov/Traces/study/?acc=SRP301630&o=acc_s%3Aa | SraRunTable(3).csv |
Shelly downloaded set 1-4 and I downloaded #5 on 1-28-2025 once we added the fifth dataset. I followed her instructions in this post.
I went to SRA Run selector webpage and used PRJNA590205 to find the associated files. I then clicked download accession list which downloads a .txt file. I copied these into an excel and saved as Cvir_prkns_dataset5_SRAids.csv. I used scp C:/Users/EmmaStrand/MyProjects/Resilience_Biomarkers_Aquaculture/Cvirg_Pmarinus_RNAseq/data/Cvir_prkns_dataset5_SRAids.csv elstrand@klone.hyak.uw.edu:/mmfs1/gscratch/scrubbed/elstrand/Cvir_disease_meta/dataset5
Custom config for nextflow: (/gscratch/scrubbed/elstrand/uw_hyak_srlab_estrand.config)
nano uw_hyak_srlab_estrand.config
### paste below information
params {
config_profile_description = 'UW Hyak Roberts labs cluster profile provided by nf-core/configs.'
config_profile_contact = 'Emma Strand emma.strand@gmgi.org'
config_profile_url = 'https://faculty.washington.edu/sr320/'
}
process {
executor = 'slurm'
queue = { task.attempt == 1 ? 'ckpt' : 'cpu-g2-mem2x' }
maxRetries = 1
clusterOptions = { "-A srlab" }
scratch = '/gscratch/scrubbed/srlab/'
resourceLimits = [
cpus: 16,
memory: '150.GB',
time: '72.h'
]
}
executor {
queuesize = 50
submitRateLimit = '1 sec'
}
singularity {
enabled = true
autoMounts = true
cacheDir = '/gscratch/scrubbed/srlab/.apptainer'
}
trace {
enabled = true
file = 'pipeline_trace.txt'
fields = 'task_id,hash,name,status,exit,submit,duration,realtime,%cpu,rss,vmem,rchar,wchar,disk,queue,hostname,cpu_model'
}
debug {
cleanup = false
}
k8s {
projectDir = '/gscratch/scrubbed/elstrand'
}
Running fetchngs after activating conda: conda activate nextflow
fetchngs.sh
#!/bin/bash
#SBATCH --job-name=fetchngs
#SBATCH --account=srlab
#SBATCH --error="%x_error.%j" #if your job fails, the error report will be put in this file
#SBATCH --output="%x_output.%j" #once your job is completed, any final job report comments will be put in this file
#SBATCH --partition=cpu-g2-mem2x
#SBATCH --nodes=1
#SBATCH --time=1-20:00:00
#SBATCH --mem=50G
#SBATCH --ntasks=7
#SBATCH --cpus-per-task=2
#SBATCH --chdir=/mmfs1/gscratch/scrubbed/elstrand/Cvir_disease_meta/dataset5
## conda environment needs to be activated prior
# run nextflow pipeline
nextflow run nf-core/fetchngs -resume \
-c /gscratch/scrubbed/elstrand/uw_hyak_srlab_estrand.config \
--input /mmfs1/gscratch/scrubbed/elstrand/Cvir_disease_meta/dataset5/Cvir_prkns_dataset5_SRAids.csv \
--outdir /mmfs1/gscratch/scrubbed/elstrand/Cvir_disease_meta/dataset5 \
--download_method sratools
After this finished ~22 min then I renamed the data files and moved output to a new fetchnegs folder.
Workflow
This workflow is fully documented in a separate post here.
Using the mamba environment I created for CGigas thermal tolerance dataset #1.
Path to fastq files: /gscratch/scrubbed/strigg/analyses/20241108_RNAseq
Path to samplesheet created by fetchNGS: /gscratch/scrubbed/strigg/analyses/20241108_RNAseq/samplesheet/samplesheet.csv
Path to Emma’s analysis: /mmfs1/gscratch/scrubbed/elstrand/Cvir_disease_2024Nov11
Path to C.virginica genome: /mmfs1/gscratch/scrubbed/elstrand/genomes/C_virginica/GCF_002022765.2_C_virginica-3.0_genomic.fna.gz
Create new samplesheet to fit rnaseq workflow:
scp xxx@klone.hyak.uw.edu:/gscratch/scrubbed/strigg/analyses/20241108_RNAseq/samplesheet/samplesheet.csv C:\Users\EmmaStrand\Downloads
scp xxx@klone.hyak.uw.edu:/gscratch/scrubbed/strigg/analyses/20241205_FetchNGS/samplesheet/samplesheet.csv C:\Users\EmmaStrand\Downloads
scp xxx@klone.hyak.uw.edu:/gscratch/scrubbed/elstrand/Cvir_disease_meta/dataset5/fetchngs/samplesheet/samplesheet.csv C:\Users\EmmaStrand\Downloads
scp C:\Users\EmmaStrand\MyProjects\Resilience_Biomarkers_Aquaculture\samplesheet_rnaseq_Cvir_disease_set1.csv xxx@klone.hyak.uw.edu:/mmfs1/gscratch/scrubbed/elstrand/Cvir_disease_2024Nov11/
scp C:\Users\EmmaStrand\MyProjects\Resilience_Biomarkers_Aquaculture\Cvirg_Pmarinus_RNAseq\data\rnaseq samplesheets\*.csv xxx@klone.hyak.uw.edu:/mmfs1/gscratch/scrubbed/elstrand/Cvir_disease_meta/samplesheets
scp C:\Users\EmmaStrand\MyProjects\Resilience_Biomarkers_Aquaculture\Cvirg_Pmarinus_RNAseq\data\rnaseq samplesheets\*.csv xxx@klone.hyak.uw.edu:/mmfs1/gscratch/scrubbed/elstrand/Cvir_disease_meta/samplesheets
scp C:\Users\EmmaStrand\MyProjects\Resilience_Biomarkers_Aquaculture\Cvirg_Pmarinus_RNAseq\data\rnaseq_samplesheets\samplesheet_rnaseq_dataset5.csv xxx@klone.hyak.uw.edu:/mmfs1/gscratch/scrubbed/elstrand/Cvir_disease_meta/samplesheets
R script on my computer to fix this sample sheet. I realized after that I over-wrote the .csv file but just moving on to uploading.
library(tidyverse)
fetchNGS_samplehseet <-
read.csv("C:/Users/EmmaStrand/MyProjects/Resilience_Biomarkers_Aquaculture/samplesheet_rnaseq_Cvir_disease_set1.csv")
rnaseq_samplesheet <- fetchNGS_samplehseet %>%
dplyr::select(sample, fastq_1, fastq_2) %>%
mutate(strandedness = "auto")
rnaseq_samplesheet %>%
write.csv("C:/Users/EmmaStrand/MyProjects/Resilience_Biomarkers_Aquaculture/samplesheet_rnaseq_Cvir_disease_set1.csv",
row.names = FALSE)
See notes after 12-4-2024 after how I used a common sbatch script to run multiple datasets.
Nextflow RNAsew workflow (01-rnaseq.sh):
Start conda environment before running sbatch.
#!/bin/bash
#SBATCH --job-name=rnaseq
#SBATCH --account=srlab
#SBATCH --error=output/"%x_error.%j" #if your job fails, the error report will be put in this file
#SBATCH --output=output/"%x_output.%j" #once your job is completed, any final job report comments will be put in this file
#SBATCH --partition=cpu-g2-mem2x
#SBATCH --nodes=1
#SBATCH --time=1-20:00:00
#SBATCH --mem=150G
#SBATCH --ntasks=7
#SBATCH --cpus-per-task=2
#SBATCH --chdir=/mmfs1/gscratch/scrubbed/elstrand/Cvir_disease_2024Nov11/scripts
#conda activate nextflow
samplesheet="/mmfs1/gscratch/scrubbed/elstrand/Cvir_disease_2024Nov11/samplesheet_rnaseq_Cvir_disease_set1.csv"
output="/mmfs1/gscratch/scrubbed/elstrand/Cvir_disease_2024Nov11/"
genome="/mmfs1/gscratch/scrubbed/elstrand/genomes/C_virginica/GCF_002022765.2_C_virginica-3.0_genomic.fna.gz"
gff="/mmfs1/gscratch/scrubbed/elstrand/genomes/C_virginica/GCF_002022765.2_C_virginica-3.0_genomic.gff.gz"
gtf="/mmfs1/gscratch/scrubbed/elstrand/genomes/C_virginica/mod_GCF_002022765.2_C_virginica-3.0_genomic.gtf.gz"
nextflow run nf-core/rnaseq \
-resume \
-profile singularity \
--input ${samplesheet} \
--outdir ${output} \
--gtf ${gtf} \
--gff ${gff} \
--fasta ${genome} \
--trimmer fastp \
--extra_fastp_args '--cut_mean_quality 30 --trim_front1 10 --trim_front2 10' \
--aligner star_salmon \
--skip_pseudo_alignment \
--multiqc_title Cvir_disease_2024Nov11 \
--deseq2_vst
2024-11-11 / 2024-11-13
First attempt to sbatch rnaseq.sh above with all four datasets at once. Should we also think about running these separately? UW was doing maintenance so checked output on 11-13. I hadn’t activated the nextflow conda environment so I activated that (conda activate nextflow) and used sbatch 01-rnaseq.sh. This started running immediately.
2024-11-14
Run failed with this error:
ERROR ~ Error executing process > 'NFCORE_RNASEQ:PREPARE_GENOME:MAKE_TRANSCRIPTS_FASTA (rsem/GCF_002022765.2_C_virginica-3.0_genomic.fna)'
Caused by:
Process `NFCORE_RNASEQ:PREPARE_GENOME:MAKE_TRANSCRIPTS_FASTA (rsem/GCF_002022765.2_C_virginica-3.0_genomic.fna)` terminated with an error exit status (255)
"rsem-extract-reference-transcripts rsem/genome 0 GCF_002022765.2_C_virginica-3.0_genomic.filtered.gtf None 0 rsem/GCF_002022765.2_C_virginica-3.0_genomic.fna" failed! Plase check if you provide correct parameters/options for the pipeline!
Stop at line : NC_007175.2 RefSeq exon 3430 3495 . + . gene_id ""; transcript_id "unknown_transcript_1"; anticodon "(pos:3461..3463)"; gbkey "tRNA"; product "tRNA-Ile"; exon_number "1";
Error Message: Attribute gene_id's value should be surrounded by double quotes and cannot be empty!
I found Sam’s post on rnaseq workflow. He documented the same issue which is how RSEM handles GTF strand parsing. So this is an issue specific to the gtf file we’re using.
Sam says: “Even though the GFF spec indicates that strand column can be one of +, -, ., or ?, RSEM only parses for + or -. And, as it turns out, our genome GFF has some . for strand info. Looking through our “merged” GenSAS GFF, it turns out there are two sets of annotations that only have . for strand info (GenSAS_5d82b316cd298-trnascan & RNAmmer-1.2). So, the decision needed to be made if we should convert these sets strands to an “artificial” value (e.g. set all of them to +), or eliminate them from the input GFF. I ended up converting GenSAS_5d82b316cd298-trnascan strand to + and eliminated RNAmmer-1.2 from the final input GFF.”
I might have an issue with . and gene_id “” that are empty? Pause to consult with Shelly. I think I can follow Sam’s notes to convert . to another character since RSEM only parses + or -, but I’m surprised that gene ids are empty here.
2024-11-20
We figured out that my issue is probably only the blank gene ID issue instead of the ‘+’/’.’ issue. Shelly ran the below code and figured out that there are only ‘+’ and ‘-‘ characters in the strand column (column #7).
(base) [strigg@klone-login03 C_virginica]$ zcat GCF_002022765.2_C_virginica-3.0_genomic.gtf.gz | awk -F"\t" '{print $7}
' | sort | uniq -c
5
761431 +
770111 -
To address the blank gene IDs issue, we changed the one gene with several exons to read gene_id=”unknown_transcript_1”
## check that changes worked
check for the changes: zcat GCF_002022765.2_C_virginica-3.0_genomic.gtf.gz | awk -F"\t" '{if($9 ~/gene_id ""/ ) gsub(/gene_id ""/,"gene_id \"unknown_transcript_1\"",$9);print $0}' | grep "unknown_t" | less
## save as new gtf file
zcat GCF_002022765.2_C_virginica-3.0_genomic.gtf.gz | awk -F"\t" '{if($9 ~/gene_id ""/ ) gsub(/gene_id ""/,"gene_id \"unknown_transcript_1\"",$9);print $0}' | gzip > mod_GCF_002022765.2_C_virginica-3.0_genomic.gtf.gz
Changing the path for the gtf file in the script above to this modified version. Started 01-rnaseq.sh
2024-11-25
The first iteration timed out so changed from 1d20h to 4d20h. Started again 11:20 am. Stopped this bc stalled at an early step.
2024-12-2
Trying this again after klone issue worked out. Bumped memory up to 150 GB instead of 100 GB. Started running right away - directory was clean of previous nextflow content beforehand.
2024-12-4
Stopped and separating each dataset.
I made a new structured in /gscratch/scrubbed/elstrand/Cvir_disease_meta that has samplesheets for each dataset and output directories for each dataset. The scripts folder has the common script and output for each dataset.
(base) [elstrand@klone-login03 Cvir_disease_meta]$ ls
dataset1 dataset2 dataset3 dataset4 samplesheets scripts
(base) [elstrand@klone-login03 samplesheets]$ ls
samplesheet_rnaseq_Cvir_disease_full.csv samplesheet_rnaseq_Cvir_disease_set2.csv samplesheet_rnaseq_Cvir_disease_set4.csv
samplesheet_rnaseq_Cvir_disease_set1.csv samplesheet_rnaseq_Cvir_disease_set3.csv
rnaseq.sh:
#!/bin/bash
#SBATCH --account=srlab
#SBATCH --error=/gscratch/scrubbed/elstrand/Cvir_disease_meta/scripts/output/"%x_error.%j" #if your job fails, the error report will be put in this file
#SBATCH --output=/gscratch/scrubbed/elstrand/Cvir_disease_meta/scripts/output/"%x_output.%j" #once your job is completed, any final job report comments will be put in this file
#SBATCH --partition=cpu-g2-mem2x
#SBATCH --nodes=1
#SBATCH --time=1-20:00:00
#SBATCH --mem=200G
#SBATCH --ntasks=7
#SBATCH --cpus-per-task=2
#conda activate nextflow
## paths defined in the sbatch command
samplesheet=$1
output=$2
multiqc_title=$3
## paths for all datasets
genome="/gscratch/scrubbed/elstrand/genomes/C_virginica/GCF_002022765.2_C_virginica-3.0_genomic.fna.gz"
gff="/gscratch/scrubbed/elstrand/genomes/C_virginica/GCF_002022765.2_C_virginica-3.0_genomic.gff.gz"
gtf="/gscratch/scrubbed/elstrand/genomes/C_virginica/mod_GCF_002022765.2_C_virginica-3.0_genomic.gtf.gz"
nextflow run nf-core/rnaseq -resume \
-c /gscratch/scrubbed/elstrand/uw_hyak_srlab_estrand.config \
-profile singularity \
--input ${samplesheet} \
--outdir ${output} \
--gtf ${gtf} \
--gff ${gff} \
--fasta ${genome} \
--trimmer fastp \
--extra_fastp_args '--cut_mean_quality 30 --trim_front1 10 --trim_front2 10' \
--aligner star_salmon \
--skip_pseudo_alignment \
--multiqc_title ${multiqc_title} \
--deseq2_vst
To run - change the samplesheet (line 1), output directory (line 2), and multiqc report name (line 3):
If cd into directory and run script from there, the work/ directory that generates go into this dataset directory rather than the scripts general directory.
conda activate nextflow
cd /gscratch/scrubbed/elstrand/Cvir_disease_meta/dataset1
sbatch -J Cvir_disease_rnaseq_dataset1 ../scripts/rnaseq.sh \
/gscratch/scrubbed/elstrand/Cvir_disease_meta/samplesheets/samplesheet_rnaseq_Cvir_disease_set1.csv \
/gscratch/scrubbed/elstrand/Cvir_disease_meta/dataset1 \
Cvir_disease_rnaseq_dataset1
12-4-2024: I started Cvir_disease_rnaseq_dataset1 ~1:45 pm. This ended up stalling so I ditched sbatch and trying to use our shared config that uses chkpt too.
12-5-2024
Starting an interactive node in a screen for dataset1 with config file from UW so it taps into ckpt resources
## start interactive
screen -S rnaseq
## (ctrl-A d to detach)
## screen -r rnaseq to reattach
## grab a node
salloc -A srlab -p cpu-g2-mem2x -N 1 -c 1 --mem=150GB --time=2-12:00:00
## start conda environment
conda activate nextflow
## navigate to output
cd /gscratch/scrubbed/elstrand/Cvir_disease_meta/dataset1
## run nextflow on dataset 1
nextflow run nf-core/rnaseq -resume \
-c /gscratch/scrubbed/elstrand/uw_hyak_srlab_estrand.config \
--input /gscratch/scrubbed/elstrand/Cvir_disease_meta/samplesheets/samplesheet_rnaseq_Cvir_disease_set1.csv \
--outdir /gscratch/scrubbed/elstrand/Cvir_disease_meta/dataset1 \
--gtf /gscratch/scrubbed/elstrand/genomes/C_virginica/mod_GCF_002022765.2_C_virginica-3.0_genomic.gtf.gz \
--gff /gscratch/scrubbed/elstrand/genomes/C_virginica/GCF_002022765.2_C_virginica-3.0_genomic.gff.gz \
--fasta /gscratch/scrubbed/elstrand/genomes/C_virginica/GCF_002022765.2_C_virginica-3.0_genomic.fna.gz \
--trimmer fastp \
--extra_fastp_args '--cut_mean_quality 30 --trim_front1 10 --trim_front2 10' \
--aligner star_salmon \
--skip_pseudo_alignment \
--multiqc_title Cvir_disease_rnaseq_dataset1 \
--deseq2_vst
Error:
ERROR ~ Error executing process > 'NFCORE_RNASEQ:PREPARE_GENOME:GUNZIP_GTF'
Caused by:
java.io.FileNotFoundException: /gscratch/scrubbed/srlab/.apptainer/.depot.galaxyproject.org-singularity-ubuntu-22.04.img.lock (Permission denied)
### also got the below
ERROR ~ Error executing process > 'NFCORE_RNASEQ:PREPARE_GENOME:GUNZIP_FASTA'
I think this is actually because of a singularity issue rather than gtf or genome file issue. I don’t know why I wouldn’t have ran into this while running an sbatch script though? I tried module load singularity/1.1.5. Shelly’s scripts don’t use a container so maybe I don’t need this? Update: didn’t fix the issue. I get the same error.
Removed -profile singularity \ and re-ran. Update: This didn’t fix the issue.
I copied the config Shelly was using and changed the directories to be my user instead and that worked!
This still stalled but we figured out that we had the wrong files downloaded with fetchNGS.
Correct files now in gscratch/scrubbed/strigg/analyses/20241205_FetchNGS
12-06-2024
With newly downloaded datasets, trying this again with the new config.
## start interactive
screen -S rnaseq
## (ctrl-A d to detach)
## screen -r rnaseq to reattach
## grab a node
salloc -A srlab -p cpu-g2-mem2x -N 1 -c 1 --mem=150GB --time=2-12:00:00
## start conda environment
conda activate nextflow
## navigate to output
cd /gscratch/scrubbed/elstrand/Cvir_disease_meta/dataset3
## run nextflow on dataset 3
nextflow run nf-core/rnaseq -resume \
-c /gscratch/scrubbed/elstrand/uw_hyak_srlab_estrand.config \
--input /gscratch/scrubbed/elstrand/Cvir_disease_meta/samplesheets/samplesheet_rnaseq_dataset3.csv \
--outdir /gscratch/scrubbed/elstrand/Cvir_disease_meta/dataset3 \
--gtf /gscratch/scrubbed/elstrand/genomes/C_virginica/mod_GCF_002022765.2_C_virginica-3.0_genomic.gtf.gz \
--gff /gscratch/scrubbed/elstrand/genomes/C_virginica/GCF_002022765.2_C_virginica-3.0_genomic.gff.gz \
--fasta /gscratch/scrubbed/elstrand/genomes/C_virginica/GCF_002022765.2_C_virginica-3.0_genomic.fna.gz \
--trimmer fastp \
--extra_fastp_args '--cut_mean_quality 30 --trim_front1 10 --trim_front2 10' \
--aligner star_salmon \
--skip_pseudo_alignment \
--multiqc_title Cvir_disease_rnaseq_dataset3 \
--deseq2_vst
This finished in 1h 52m 56s 12-6-2024!
Transferring data to Gannet:
rsync --archive --verbose --progress multiqc/star_salmon/multiqc_report.html emma.strand@gannet.fish.washington.edu:/volume2/web/emma.strand/rnaseq/Cvir_Prkns_rnaseq_dataset3
rsync --archive --verbose --progress pipeline_info/ emma.strand@gannet.fish.washington.edu:/volume2/web/emma.strand/rnaseq/Cvir_Prkns_rnaseq_dataset3
rsync --archive --verbose --progress pipeline_trace.txt emma.strand@gannet.fish.washington.edu:/volume2/web/emma.strand/rnaseq/Cvir_Prkns_rnaseq_dataset3
rsync --archive --verbose --progress pipeline_trace.txt emma.strand@gannet.fish.washington.edu:/volume2/web/emma.strand/rnaseq/Cvir_Prkns_rnaseq_dataset3
rsync --archive --verbose --progress star_salmon/deseq2_qc/ emma.strand@gannet.fish.washington.edu:/volume2/web/emma.strand/rnaseq/Cvir_Prkns_rnaseq_dataset3
rsync --archive --verbose --progress log/ emma.strand@gannet.fish.washington.edu:/volume2/web/emma.strand/rnaseq/Cvir_Prkns_rnaseq_dataset3
rsync --archive --verbose --progress *.tsv emma.strand@gannet.fish.washington.edu:/volume2/web/emma.strand/rnaseq/Cvir_Prkns_rnaseq_dataset3
I realized that we aren’t including the correct flag to fully analyze novel splice variants / de novo transcripts. Need to add the below flags (come back to filling this in)
--extra_star_align_args ''
Explanation of 2-pass to get novel transcripts:
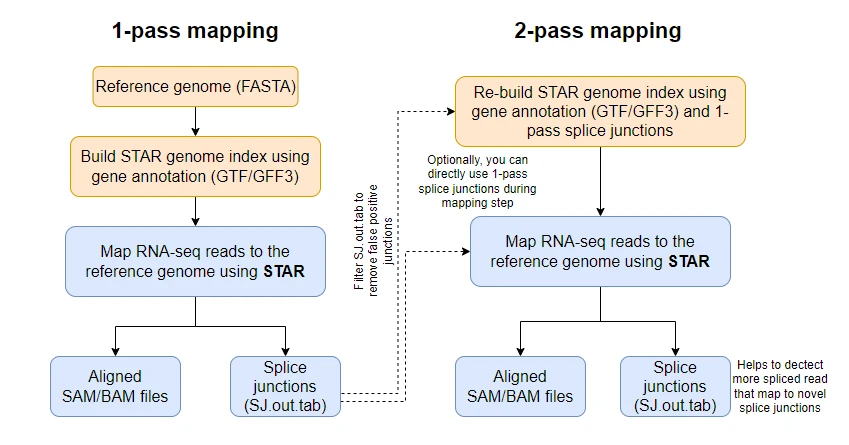
Running dataset 1 but trying with sbatch script and config file. Added -c /gscratch/scrubbed/elstrand/uw_hyak_srlab_estrand.config to the sbatch script.
conda activate nextflow
cd /gscratch/scrubbed/elstrand/Cvir_disease_meta/dataset1
sbatch -J Cvir_disease_rnaseq_dataset1 ../scripts/rnaseq.sh \
/gscratch/scrubbed/elstrand/Cvir_disease_meta/samplesheets/samplesheet_rnaseq_dataset1.csv \
/gscratch/scrubbed/elstrand/Cvir_disease_meta/dataset1 \
Cvir_disease_rnaseq_dataset1
This worked for now! It’s running so I will continue to check in on the output file: /gscratch/scrubbed/elstrand/Cvir_disease_meta/scripts/output/Cvir_disease_rnaseq_dataset1_output.22760577
Running dataset 2 with sbatch script with config file.
conda activate nextflow
cd /gscratch/scrubbed/elstrand/Cvir_disease_meta/dataset2
sbatch -J Cvir_disease_rnaseq_dataset2 ../scripts/rnaseq.sh \
/gscratch/scrubbed/elstrand/Cvir_disease_meta/samplesheets/samplesheet_rnaseq_dataset2.csv \
/gscratch/scrubbed/elstrand/Cvir_disease_meta/dataset2 \
Cvir_disease_rnaseq_dataset2
Running dataset 4 with sbatch script with config file.
conda activate nextflow
cd /gscratch/scrubbed/elstrand/Cvir_disease_meta/dataset4
sbatch -J Cvir_disease_rnaseq_dataset4 ../scripts/rnaseq.sh \
/gscratch/scrubbed/elstrand/Cvir_disease_meta/samplesheets/samplesheet_rnaseq_dataset4.csv \
/gscratch/scrubbed/elstrand/Cvir_disease_meta/dataset4 \
Cvir_disease_rnaseq_dataset4
12-9-2024
Datasets 2 and 4 finished but dataset1 was killed b/c of a time limit. restarting this script.
Transferring data to Gannet:
rsync --archive --verbose --progress multiqc/star_salmon/multiqc_report.html emma.strand@gannet.fish.washington.edu:/volume2/web/emma.strand/rnaseq/Cvir_Prkns_rnaseq_dataset2/pipeline_info
rsync --archive --verbose --progress pipeline_info/ emma.strand@gannet.fish.washington.edu:/volume2/web/emma.strand/rnaseq/Cvir_Prkns_rnaseq_dataset2/pipeline_info
rsync --archive --verbose --progress pipeline_trace.txt emma.strand@gannet.fish.washington.edu:/volume2/web/emma.strand/rnaseq/Cvir_Prkns_rnaseq_dataset2/pipeline_info
rsync --archive --verbose --progress star_salmon/deseq2_qc/ emma.strand@gannet.fish.washington.edu:/volume2/web/emma.strand/rnaseq/Cvir_Prkns_rnaseq_dataset2/deseq2_qc
rsync --archive --verbose --progress star_salmon/log/ emma.strand@gannet.fish.washington.edu:/volume2/web/emma.strand/rnaseq/Cvir_Prkns_rnaseq_dataset2/log
rsync --archive --verbose --progress star_salmon/*.tsv emma.strand@gannet.fish.washington.edu:/volume2/web/emma.strand/rnaseq/Cvir_Prkns_rnaseq_dataset2
rsync --archive --verbose --progress multiqc/star_salmon/multiqc_report.html emma.strand@gannet.fish.washington.edu:/volume2/web/emma.strand/rnaseq/Cvir_Prkns_rnaseq_dataset4/pipeline_info
rsync --archive --verbose --progress pipeline_info/ emma.strand@gannet.fish.washington.edu:/volume2/web/emma.strand/rnaseq/Cvir_Prkns_rnaseq_dataset4/pipeline_info
rsync --archive --verbose --progress pipeline_trace.txt emma.strand@gannet.fish.washington.edu:/volume2/web/emma.strand/rnaseq/Cvir_Prkns_rnaseq_dataset4/pipeline_info
rsync --archive --verbose --progress star_salmon/deseq2_qc/ emma.strand@gannet.fish.washington.edu:/volume2/web/emma.strand/rnaseq/Cvir_Prkns_rnaseq_dataset4/deseq2_qc
rsync --archive --verbose --progress star_salmon/log/ emma.strand@gannet.fish.washington.edu:/volume2/web/emma.strand/rnaseq/Cvir_Prkns_rnaseq_dataset4/log
rsync --archive --verbose --progress star_salmon/*.tsv emma.strand@gannet.fish.washington.edu:/volume2/web/emma.strand/rnaseq/Cvir_Prkns_rnaseq_dataset4
Rm dataset1 repo and make new one – wasn’t sure if rnaseq pipeline stopped mid sample b/c of time limit so don’t want to risk it.
conda activate nextflow
cd /gscratch/scrubbed/elstrand/Cvir_disease_meta/dataset1
sbatch -J Cvir_disease_rnaseq_dataset1 ../scripts/rnaseq.sh \
/gscratch/scrubbed/elstrand/Cvir_disease_meta/samplesheets/samplesheet_rnaseq_dataset1.csv \
/gscratch/scrubbed/elstrand/Cvir_disease_meta/dataset1 \
Cvir_disease_rnaseq_dataset1
Re-started at ~1:30 pm after I changed the time limit to 4d-20 hr.. It was 1d-20hr so are these files just really big and need more than 2 days? That would be surprising..
2024-12-11
This ran in 5 hr! Not sure what happened the first time around.
Moving data to Gannet.
rsync --archive --verbose --progress multiqc/star_salmon/multiqc_report.html emma.strand@gannet.fish.washington.edu:/volume2/web/emma.strand/rnaseq/Cvir_Prkns_rnaseq_dataset1/pipeline_info
rsync --archive --verbose --progress pipeline_info/ emma.strand@gannet.fish.washington.edu:/volume2/web/emma.strand/rnaseq/Cvir_Prkns_rnaseq_dataset1/pipeline_info
rsync --archive --verbose --progress pipeline_trace.txt emma.strand@gannet.fish.washington.edu:/volume2/web/emma.strand/rnaseq/Cvir_Prkns_rnaseq_dataset1/pipeline_info
rsync --archive --verbose --progress star_salmon/deseq2_qc/ emma.strand@gannet.fish.washington.edu:/volume2/web/emma.strand/rnaseq/Cvir_Prkns_rnaseq_dataset1/deseq2_qc
rsync --archive --verbose --progress star_salmon/log/ emma.strand@gannet.fish.washington.edu:/volume2/web/emma.strand/rnaseq/Cvir_Prkns_rnaseq_dataset1/log
rsync --archive --verbose --progress star_salmon/*.tsv emma.strand@gannet.fish.washington.edu:/volume2/web/emma.strand/rnaseq/Cvir_Prkns_rnaseq_dataset1
Now moving onto interpretation of this data via differential abundance workflow and/or custom script in R.
Running Cgigas from dataset 3 separately so the reference is Cgigas. I copied the rnaseq script and changed the genome paths.
scp C:\Users\EmmaStrand\MyProjects\Resilience_Biomarkers_Aquaculture\Cvirg_Pmarinus_RNAseq\data\rnaseq_samplesheets\samplesheet_rnaseq_dataset3_Cgigas.csv xxx@klone.hyak.uw.edu:/mmfs1/gscratch/scrubbed/elstrand/Cvir_disease_meta/samplesheets
genome=”/mmfs1/gscratch/scrubbed/elstrand/genomes/C_gigas/GCF_963853765.1_xbMagGiga1.1_genomic.fna.gz” gff=”/mmfs1/gscratch/scrubbed/elstrand/genomes/C_gigas/GCF_963853765.1_xbMagGiga1.1_genomic.gff.gz” gtf=”/mmfs1/gscratch/scrubbed/elstrand/genomes/C_gigas/GCF_963853765.1_xbMagGiga1.1_genomic.gtf.gz”
conda activate nextflow
cd /gscratch/scrubbed/elstrand/Cvir_disease_meta/dataset3_Cgigas
sbatch -J Cvir_disease_rnaseq_dataset3_Cgigas ../scripts/rnaseq_Cgigas.sh \
/gscratch/scrubbed/elstrand/Cvir_disease_meta/samplesheets/samplesheet_rnaseq_dataset3_Cgigas.csv \
/gscratch/scrubbed/elstrand/Cvir_disease_meta/dataset3_Cgigas \
Cvir_disease_rnaseq_dataset3_Cgigas
2024-12-13
This ran in 16 mintues! That is so fast.. double check on multiqc that everything ran..
rsync --archive --verbose --progress multiqc/star_salmon/multiqc_report.html emma.strand@gannet.fish.washington.edu:/volume2/web/emma.strand/rnaseq/Cvir_Prkns_rnaseq_dataset3_Cgigas/pipeline_info
rsync --archive --verbose --progress pipeline_info/ emma.strand@gannet.fish.washington.edu:/volume2/web/emma.strand/rnaseq/Cvir_Prkns_rnaseq_dataset3_Cgigas/pipeline_info
rsync --archive --verbose --progress pipeline_trace.txt emma.strand@gannet.fish.washington.edu:/volume2/web/emma.strand/rnaseq/Cvir_Prkns_rnaseq_dataset3_Cgigas/pipeline_info
rsync --archive --verbose --progress star_salmon/deseq2_qc/ emma.strand@gannet.fish.washington.edu:/volume2/web/emma.strand/rnaseq/Cvir_Prkns_rnaseq_dataset3_Cgigas/deseq2_qc
rsync --archive --verbose --progress star_salmon/log/ emma.strand@gannet.fish.washington.edu:/volume2/web/emma.strand/rnaseq/Cvir_Prkns_rnaseq_dataset3_Cgigas/log
rsync --archive --verbose --progress star_salmon/*.tsv emma.strand@gannet.fish.washington.edu:/volume2/web/emma.strand/rnaseq/Cvir_Prkns_rnaseq_dataset3_Cgigas
rsync --archive --verbose --progress ../scripts/output/Cvir_disease_rnaseq_dataset3_Cgigas_* emma.strand@gannet.fish.washington.edu:/volume2/web/emma.strand/rnaseq/Cvir_Prkns_rnaseq_dataset3_Cgigas
Steve was getting this warning:
WARNING: you specified the options for cutting by quality, but forogt to enable any of cut_front/cut_tail/cut_right. This will have no effect.
Running Cvir_Prkns_rnaseq_dataset3_Cgigas in srun to see if I also get this warning and it doesn’t show up in my output files..
## start interactive
screen -S rnaseq
## (ctrl-A d to detach)
## screen -r rnaseq to reattach
## screen -X -S [session # you want to kill] kill
## grab a node
salloc -A srlab -p cpu-g2-mem2x -N 1 -c 1 --mem=150GB --time=2-12:00:00
## start conda environment
conda activate nextflow
## navigate to output
cd /gscratch/scrubbed/elstrand/Cvir_disease_meta/dataset3_Cgigas
## run nextflow on dataset 3
nextflow run nf-core/rnaseq -resume \
-c /gscratch/scrubbed/elstrand/uw_hyak_srlab_estrand.config \
--input /gscratch/scrubbed/elstrand/Cvir_disease_meta/samplesheets/samplesheet_rnaseq_dataset3_Cgigas.csv \
--outdir /gscratch/scrubbed/elstrand/Cvir_disease_meta/dataset3_Cgigas \
--gtf /gscratch/scrubbed/elstrand/genomes/C_gigas/GCF_963853765.1_xbMagGiga1.1_genomic.gtf.gz \
--gff /gscratch/scrubbed/elstrand/genomes/C_gigas/GCF_963853765.1_xbMagGiga1.1_genomic.gff.gz \
--fasta /gscratch/scrubbed/elstrand/genomes/C_gigas/GCF_963853765.1_xbMagGiga1.1_genomic.fna.gz \
--trimmer fastp \
--extra_fastp_args '--cut_mean_quality 30 --trim_front1 10 --trim_front2 10' \
--aligner star_salmon \
--skip_pseudo_alignment \
--multiqc_title Cvir_disease_rnaseq_dataset3_Cgigas \
--deseq2_vst
I still can’t find this warning.. Maybe it’s a version issue?
Checking rnaseq workflow outputs via multiqc
Notes:
- Re-run dataset3 b/c the trimming is too large compared to the length of the sequences (cutting 10 off 50 bp long)
Re-run dataset3 with new parameters
I first changed parameters in the rnaseq file to be trimming defined by sbatch call. Then I moved the Cgigas data, samplesheet and script to its own folder.
Cgigas dataset3:
Only ‘–cut_mean_quality 30’ without the trim parameters
conda activate nextflow
cd /gscratch/scrubbed/elstrand/Cgigas_disease_meta/dataset3_Cgigas
sbatch -J Cgigas_disease_rnaseq_dataset3 ../scripts/rnaseq_Cgigas.sh \
/gscratch/scrubbed/elstrand/Cgigas_disease_meta/samplesheets/samplesheet_rnaseq_dataset3_Cgigas.csv \
/gscratch/scrubbed/elstrand/Cgigas_disease_meta/dataset3_Cgigas \
Cgigas_disease_rnaseq_dataset3
Cvir dataset3:
I then changed the samplesheet on github to reflect Cvir only instead of both and scp C:\Users\EmmaStrand\MyProjects\Resilience_Biomarkers_Aquaculture\Cvirg_Pmarinus_RNAseq\data\rnaseq_samplesheets\samplesheet_rnaseq_dataset3.csv xxx@klone.hyak.uw.edu:/mmfs1/gscratch/scrubbed/elstrand/Cvir_disease_meta/samplesheets
Only ‘–cut_mean_quality 30’ without the trim parameters
conda activate nextflow
cd \gscratch\scrubbed\elstrand\Cvir_disease_meta\dataset3
sbatch -J Cvir_disease_rnaseq_dataset3 ../scripts/rnaseq_Cvir.sh \
/gscratch/scrubbed/elstrand/Cvir_disease_meta/samplesheets/samplesheet_rnaseq_dataset3.csv \
/gscratch/scrubbed/elstrand/Cvir_disease_meta/dataset3 \
Cvir_disease_rnaseq_dataset3
Running dataset 5
1-28-2025 - 1-29-2025
If re-running this, remove the pipeline_trace.txt file. It won’t re-run if there is already an existing file
conda activate nextflow
cd /gscratch/scrubbed/elstrand/Cvir_disease_meta/dataset5/rnaseq
sbatch -J Cvir_disease_rnaseq_dataset5 ../../scripts/rnaseq_Cvir.sh \
/gscratch/scrubbed/elstrand/Cvir_disease_meta/dataset5/rnaseq/samplesheet_rnaseq_dataset5.csv \
/gscratch/scrubbed/elstrand/Cvir_disease_meta/dataset5 \
Cvir_disease_rnaseq_dataset5 \
'--cut_mean_quality 30 --trim_front1 10 --trim_front2 10'
I ran into this error. Figured out that I needed ${fastp} in the script rather than $4 in the actual nextflow code. Changed that and re-ran, but still getting this same error even with ${fastp}..
WARN: The following invalid input values have been detected:
* --cut_mean_quality: 30
* --trim_front1: 10
* --trim_front2: 10
ERROR ~ Validation of pipeline parameters failed!
Moved '--cut_mean_quality 30 --trim_front1 10 --trim_front2 10' to the slurm script and ran again with this embedded in nextflow.
conda activate nextflow
cd /gscratch/scrubbed/elstrand/Cvir_disease_meta/dataset5/rnaseq
sbatch -J Cvir_disease_rnaseq_dataset5 ../../scripts/rnaseq_Cvir.sh \
/gscratch/scrubbed/elstrand/Cvir_disease_meta/dataset5/samplesheet_rnaseq_dataset5.csv \
/gscratch/scrubbed/elstrand/Cvir_disease_meta/dataset5 \
Cvir_disease_rnaseq_dataset5
I’m getting this error: ERROR ~ Validation of pipeline parameters failed! but no mention of which parameters. This is because I need to modify the gtf file so did that again and re-ran the script. I STILL get the parameters issue. Cannot validate pipeline parameters.. Is the config file wrong? I changed the paths of the .apptainer back to srlab to mimic Shelly’s and only changing the directory at the bottom of the file. Re-ran script. I realized rnaseq/ was in the samplesheet path when it shouldn’t have been. Fixed and re-ran. Still got this issue.. Removed the srlab directories from my config file so it’s all directed to my user on scrubbed. Tried again.
Got this error:
* --input (/gscratch/scrubbed/elstrand/Cvir_disease_meta/dataset5/samplesheet_rnaseq_dataset5.csv): Validation of file failed:
-> Entry 1: Error for field 'fastq_2' (/mmfs1/gscratch/scrubbed/elstrand/Cvir_disease_meta/dataset5/fastq/SRX7172518_SRR10482877_2.fastq.gz): FastQ file for reads 2 cannot contain spaces and must have extension '.fq.gz' or '.fastq.gz'
I had changed the fastq folder to raw_data but fastq is in the path in the samplesheet. So I changed that folder back. Re-ran again and it worked!!
This output was in dataset5 and then afterwards I moved this to rnaseq. This dataset took 3 hours to complete.
Moving to gannet:
rsync --archive --verbose --progress multiqc/star_salmon/multiqc_report.html emma.strand@gannet.fish.washington.edu:/volume2/web/emma.strand/rnaseq/Cvir_Prkns_rnaseq_dataset5/pipeline_info
rsync --archive --verbose --progress pipeline_info/ emma.strand@gannet.fish.washington.edu:/volume2/web/emma.strand/rnaseq/Cvir_Prkns_rnaseq_dataset5/pipeline_info
rsync --archive --verbose --progress pipeline_trace.txt emma.strand@gannet.fish.washington.edu:/volume2/web/emma.strand/rnaseq/Cvir_Prkns_rnaseq_dataset5/pipeline_info
rsync --archive --verbose --progress star_salmon/deseq2_qc/ emma.strand@gannet.fish.washington.edu:/volume2/web/emma.strand/rnaseq/Cvir_Prkns_rnaseq_dataset5/deseq2_qc
rsync --archive --verbose --progress star_salmon/log/ emma.strand@gannet.fish.washington.edu:/volume2/web/emma.strand/rnaseq/Cvir_Prkns_rnaseq_dataset5/log
cd \gscratch\scrubbed\elstrand\Cvir_disease_meta\dataset5\rnaseq\star_salmon
rsync --archive --verbose --progress *.tsv emma.strand@gannet.fish.washington.edu:/volume2/web/emma.strand/rnaseq/Cvir_Prkns_rnaseq_dataset5
cd \gscratch\scrubbed\elstrand\Cvir_disease_meta\scripts\output
rsync --archive --verbose --progress * emma.strand@gannet.fish.washington.edu:/volume2/web/emma.strand/rnaseq/Cvir_Prkns_rnaseq_dataset5
Moved genomes, scripts, and config file to /gscratch/srlab/elstrand