ran on 11-04-2024
salloc -A srlab -p cpu-g2-mem2x -N 1 -c 1 --mem=100GB --time=2-12:00:00
mamba activate nextflow
nextflow run nf-core/denovotranscript \
-c /gscratch/srlab/strigg/bin/uw_hyak_srlab.config \
--input /gscratch/scrubbed/strigg/analyses/20240925/samplesheet/samplesheet.csv \
--outdir /gscratch/scrubbed/strigg/analyses/20241104_denovo \
--extra_fastp_args='--trim_front1 10 --trim_front2 10' \
--remove_ribo_rna \
--assemblers rnaspades \
-resume \
-with-report nf_report.html \
-with-trace \
-with-timeline nf_timeline.html
This errored out at SORTMERNA step and couldn’t figure out why. Here are some screenshots of what was happening:


I reran the pipeline in a new screen session and node allocation.
the pipeline errored out again. I submitted an issue via github. it seems like a memory issue. Asking for too much memory that is beyond what is allocated
2024-11-08
I reran the pipeline in a new screen session and new directory
nextflow run nf-core/denovotranscript \
-c /gscratch/srlab/strigg/bin/uw_hyak_srlab.config \
--input /gscratch/scrubbed/strigg/analyses/20240925/samplesheet/samplesheet.csv \
--outdir /gscratch/scrubbed/strigg/analyses/20241108_denovo \
--extra_fastp_args='--trim_front1 10 --trim_front2 10' \
--remove_ribo_rna \
--assemblers rnaspades \
-resume \
-with-report nf_report.html \
-with-timeline nf_timeline.html
It again stalled out at rnaSPADES. I think because of a memory issue.
2024-11-09
I reduced the number of samples to do the assembly and just selected one sample per family and timepoint. The criteria for selection was highest number of reads after adapter and quality filtering (taken from the general statistics data generated by multiqc from Emma’s RNAseq).
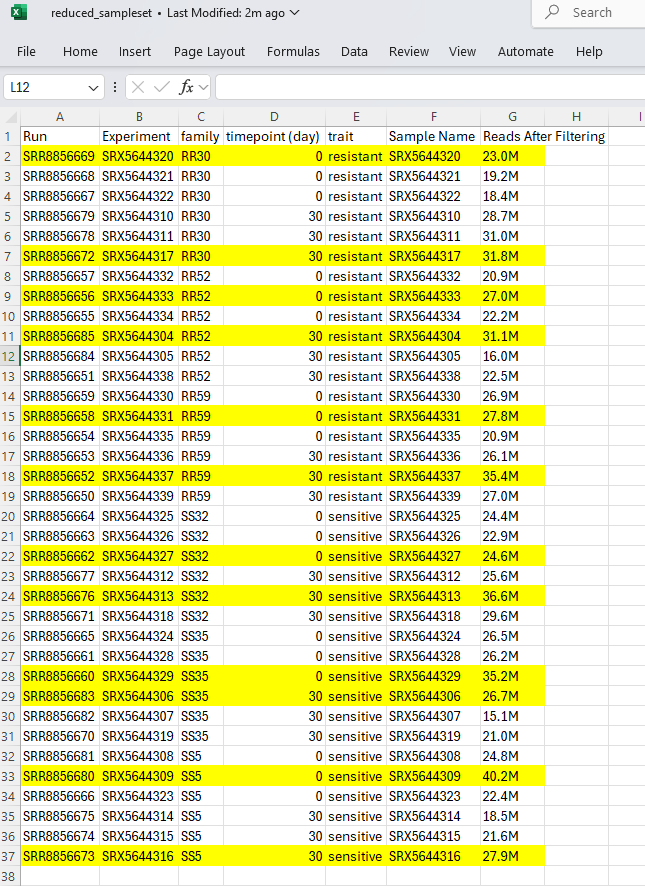
I created a list of the experiment ids using vim:
SRX5644320
SRX5644317
SRX5644333
SRX5644304
SRX5644331
SRX5644337
SRX5644327
SRX5644313
SRX5644329
SRX5644306
SRX5644309
SRX5644316
:wq ids.csv
I created a new samplesheet for these 12 samples:
#use grep to search for the ids in ids.csv in samplesheet.csv and save that as a new file
[strigg@klone-login03 20241109_denovo]$ grep -Ff ids.csv ../20240925/samplesheet/samplesheet.csv > samplesheet.csv
#add header to new samplesheet
[strigg@klone-login03 20241109_denovo]$ head -1 ../20240925/samplesheet/samplesheet.csv | cat - samplesheet.csv > samplesheet2.csv
#remove samplesheet that doesn't have header
[strigg@klone-login03 20241109_denovo]$ rm samplesheet.csv
I reran the pipeline
nextflow run nf-core/denovotranscript \
-c /gscratch/srlab/strigg/bin/uw_hyak_srlab.config \
--input /gscratch/scrubbed/strigg/analyses/20241109_denovo/samplesheet2.csv \
--outdir /gscratch/scrubbed/strigg/analyses/20241109_denovo \
--extra_fastp_args='--trim_front1 10 --trim_front2 10' \
--remove_ribo_rna \
--assemblers rnaspades \
-with-report nf_report.html \
-with-timeline nf_timeline.html