This entry describes how I set up nextflow on the UW HPC Klone server and got fetchngs to run successfully
Following the guide outlined by Carson Miller in the UW Peds Dept I did the following:
1. Install mamba and create a mamba environment containing nextflow and nf-core.
NOTE: This was a one-time step and does not need to be repeated. This same nextflow environment can be used by anyone to run any nextflow pipeline
Ran the following lines of code from my home directory /mmfs1/home/strigg
wget https://github.com/conda-forge/miniforge/releases/latest/download/Mambaforge-Linux-x86_64.sh
bash Mambaforge-Linux-x86_64.sh -b -p $HOME/mambaforge
mamba create -n nextflow -c bioconda -c conda-forge python=3.8 nf-core nextflow -y -q
2. Create .config file for srlab
NOTE: This was a one-time step and does not need to be repeated. This same config file can be used by anyone to run any nextflow pipeline.
/gscratch/srlab/strigg/bin/uw_hyak_srlab.config
params {
config_profile_description = 'UW Hyak Roberts labs cluster profile provided by nf-core/configs.'
config_profile_contact = 'Shelly A. Wanamaker @shellywanamaker'
config_profile_url = 'https://faculty.washington.edu/sr320/'
max_memory = 742.GB
max_cpus = 40
max_time = 72.h
}
process {
executor = 'slurm'
queue = { task.attempt == 1 ? 'ckpt' : 'cpu-g2-mem2x' }
maxRetries = 1
clusterOptions = { "-A srlab" }
scratch = '/gscratch/scrubbed/srlab/'
}
executor {
queuesize = 50
submitRateLimit = '1 sec'
}
singularity {
enabled = true
autoMounts = true
cacheDir = '/gscratch/scrubbed/srlab/.apptainer'
}
debug {
cleanup = false
}
3. Run nextflow on a compute node.
NOTE: The reason Carson prefers using screen as opposed to SLURM is because (particularly when testing) it enables more interactivity. If you want to cancel a run you simply attach to the screen, Ctrl + C and it will cancel the head job as well as all other running jobs. If you try to cancel a SLURM script, you will have to run another script to cancel all the jobs running on compute nodes.
Everything in the code below is generalizable to nextflow nf-core pipelines. The ‘
# create a screen session
screen -S nextflow
# request a compute node (mem and time requests can be modified)
salloc -A srlab -p cpu-g2-mem2x -N 1 -c 1 --mem=16GB --time=12:00:00
# load the nextflow environment
mamba activate nextflow
# run nextflow pipeline
nextflow run \
nf-core/<pipeline> \
-resume \
-c /gscratch/srlab/strigg/bin/uw_hyak_srlab.config \
<pipeline specific flags>
Running fetchngs
# create a screen session
screen -S nextflow
# request a compute node (mem and time requests can be modified)
salloc -A srlab -p cpu-g2-mem2x -N 1 -c 1 --mem=16GB --time=12:00:00
# load the nextflow environment
mamba activate nextflow
# run nextflow pipeline
nextflow run \
nf-core/fetchngs \
-resume \
-c /gscratch/srlab/strigg/bin/uw_hyak_srlab.config \
--input /gscratch/scrubbed/strigg/analyses/20240925/ids.csv \
--outdir /gscratch/scrubbed/strigg/analyses/20240925 \
--download_method sratools
Pipeline-specific parameters explained:
--input /gscratch/scrubbed/strigg/analyses/20240925/ids.csv
fetchngs requires an input file containing the SRA run IDs. To create this, I got the list of run IDs from Roberto’s C. gigas thermotolerance study and dataset from SRA here. I copied and pasted the values from this table into an excel sheet. I then copied just the run ID column and pasted the values into a plain text file. I did this because copying and pasting directly from the excel sheet into vim I think led to hidden spaces or characters in the file because fetchngs wouldn’t complete and complained there was a mix of IDs. Coping and pasting from the plain text file into vim solved the problem. The file I created was called ids.csv
SRR8856650
SRR8856651
SRR8856652
SRR8856653
SRR8856654
SRR8856655
SRR8856656
SRR8856657
SRR8856658
SRR8856659
SRR8856660
SRR8856661
SRR8856662
SRR8856663
SRR8856664
SRR8856665
SRR8856666
SRR8856667
SRR8856668
SRR8856669
SRR8856670
SRR8856671
SRR8856672
SRR8856673
SRR8856674
SRR8856675
SRR8856676
SRR8856677
SRR8856678
SRR8856679
SRR8856680
SRR8856681
SRR8856682
SRR8856683
SRR8856684
SRR8856685
The parameter below specifies the output directory
--outdir /gscratch/scrubbed/strigg/analyses/20240925
The parameter below specifies which download method to use
--download_method sratools
I used this flag because I wasn’t sure the FTP was working. This was before I realized there were hidden characters from copying and pasting from excel into vim (see above). The usage documentation states “ If you are having issues and prefer to use sra-tools or Aspera instead, you can set the –download_method parameter to –download_method sratools or –download_method aspera, respectively”
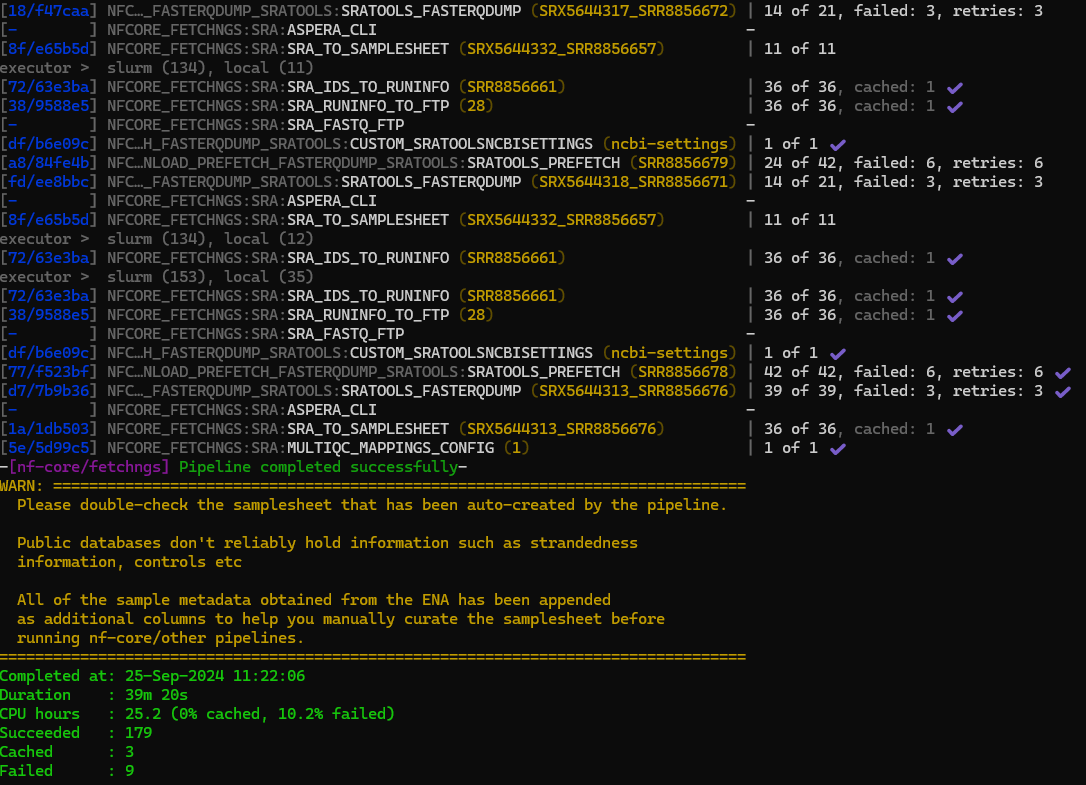
fetchngs output
The pipeline completed in 39 minutes
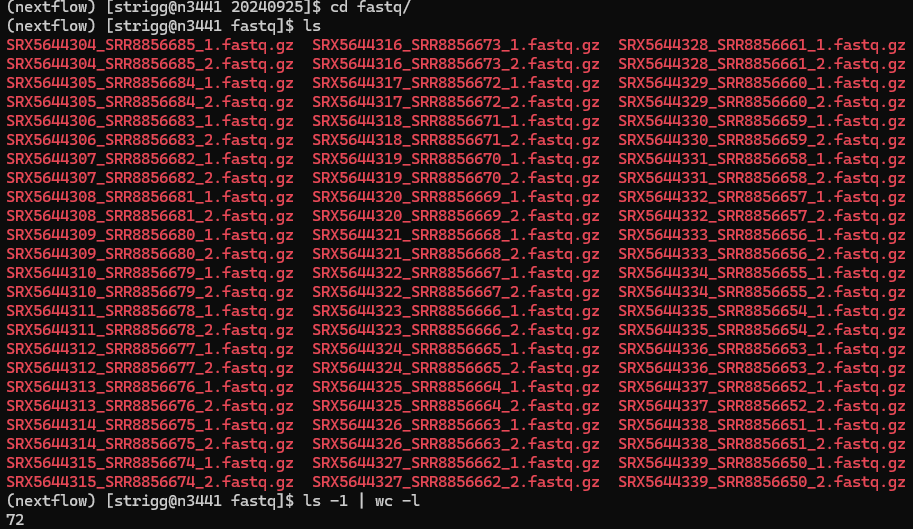
The data is paired end so there are 72 files, 2 for each of the 36 tissue samples.
The pipeline created a samplesheet.csv that can be used in other nf-core pipelines: