This entry describes running denovotranscript on the UW HPC Klone server. I generally followed the example workflow provided in the denovotranscript usage documentation.
It’s going to use the samplesheet.csv as input that was created by fetchngs
1. Run the pipeline with –qc_only with default params to check the quality of your reads.
# create a screen session
screen -S nextflow
# request a compute node (mem and time requests can be modified)
salloc -A srlab -p cpu-g2-mem2x -N 1 -c 1 --mem=16GB --time=12:00:00
# load the nextflow environment
mamba activate nextflow
# run nextflow pipeline
nextflow run \
nf-core/denovotranscript \
-resume \
-c /gscratch/srlab/strigg/bin/uw_hyak_srlab.config \
--input /gscratch/scrubbed/strigg/analyses/20240925/samplesheet/samplesheet.csv \
--outdir /gscratch/scrubbed/strigg/analyses/20240925_denovo \
--qc_only
This completed successfully
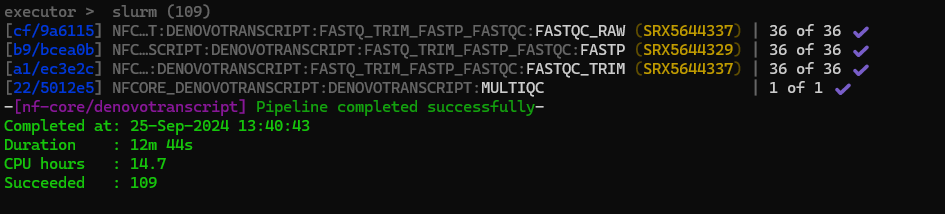
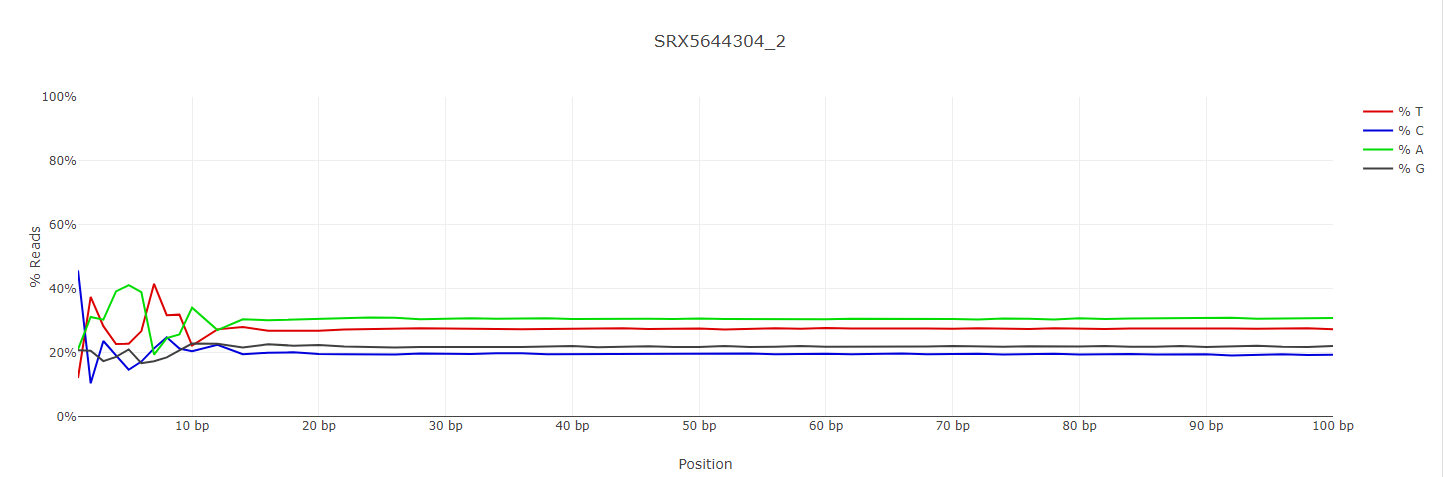
I looked at the multiqc and decided the first 10 bases should be trimmed from all reads. Example below
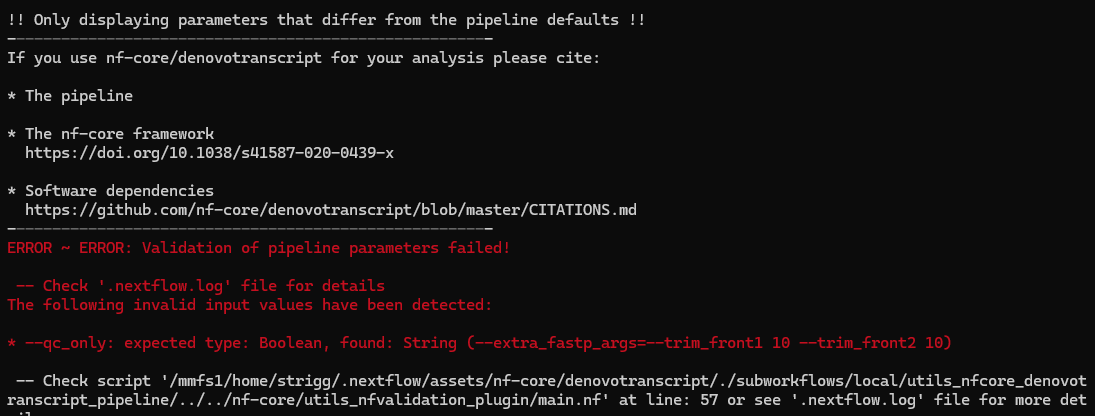
- Run the pipeline with qc_only and any custom parameters that you have decided to use based on your data. Use resume to avoid unnecessarily rerunning unchanged steps. I next ran:
nextflow run nf-core/denovotranscript \ -c /gscratch/srlab/strigg/bin/uw_hyak_srlab.config \ --input /gscratch/scrubbed/strigg/analyses/20240925/samplesheet/samplesheet.csv \ --outdir /gscratch/scrubbed/strigg/analyses/20240925_denovo \ --qc_only --extra_fastp_args='--trim_front1 10 --trim_front2 10' \ --remove_ribo_rna \ -resumethis failed.
 I closed the screen with ctrl + A + D and then screen -XS nextflow quit
I closed the screen with ctrl + A + D and then screen -XS nextflow quit
I decided to just try the whole pipeline and ran:
screen -S nextflow
salloc -A srlab -p cpu-g2-mem2x -N 1 -c 1 --mem=100GB --time=2-12:00:00
mamba activate nextflow
nextflow run nf-core/denovotranscript \
-c /gscratch/srlab/strigg/bin/uw_hyak_srlab.config \
--input /gscratch/scrubbed/strigg/analyses/20240925/samplesheet/samplesheet.csv \
--outdir /gscratch/scrubbed/strigg/analyses/20240925_denovo \
--extra_fastp_args='--trim_front1 10 --trim_front2 10' \
--remove_ribo_rna \
--busco_lineage= 'mollusca_odb10' \
-resume
Next time I would like to run the nextflow code with the following parameters:
-with-report nf_report
-with-trace
-with-timeline nf_timeline
Will see how far things get tomorrow.